Tutorial - Analyzing a phased trio
Introduction
In some scenarios, it can be useful to gather genome sequencing data from both the parents and offspring, this can form a "trio" (e.g. mom, dad, and child)
After doing variant calling and producing a VCF file, the VCF data can additionally be "phased" which tells us information about each "haplotype" of a VCF dataset
For this tutorial, we will not go through the process of creating such a file, but will look at trio from the 1000 genomes dataset for a Kinh-Vietnamese trio (KHV in filename)
- VCF tabix file https://hgdownload.soe.ucsc.edu/gbdb/hg38/1000Genomes/trio/HG02024_VN049_KHV/HG02024_VN049_KHVTrio.chr1.vcf.gz
- VCF tabix index file https://hgdownload.soe.ucsc.edu/gbdb/hg38/1000Genomes/trio/HG02024_VN049_KHV/HG02024_VN049_KHVTrio.chr1.vcf.gz
You can either use the JBrowse CLI or GUI to add this file to JBrowse. Note that this file only represents chr1
After adding the VCF it will look like this
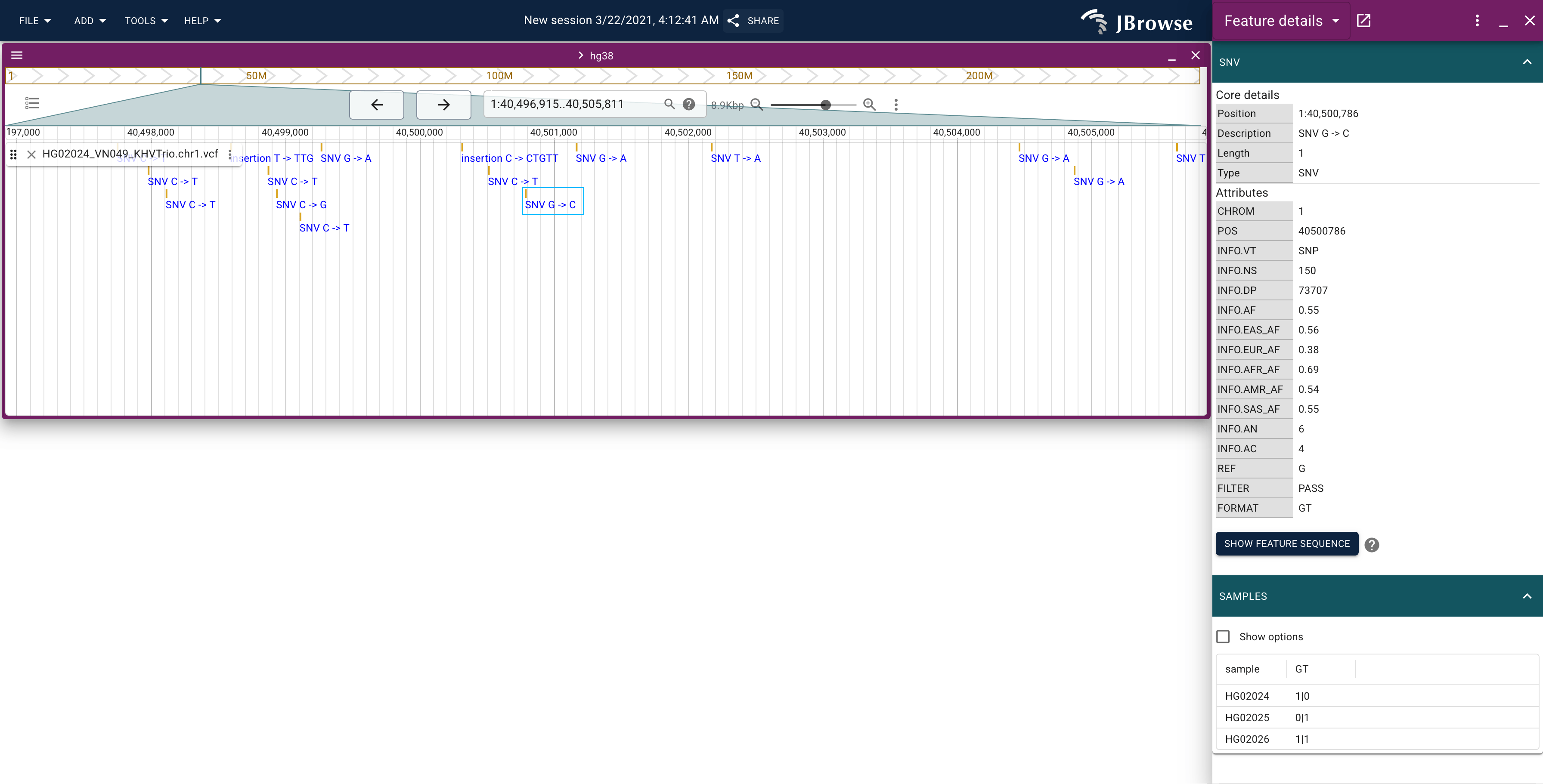
Enabling the matrix view
JBrowse can show variant datasets using a specialized "display mode" called the "Multi-sample variant display (matrix)"
This mode produces a new visualization modality that shows all the samples at once, along with all the variants, in a dense "heatmap" or "matrix" style display
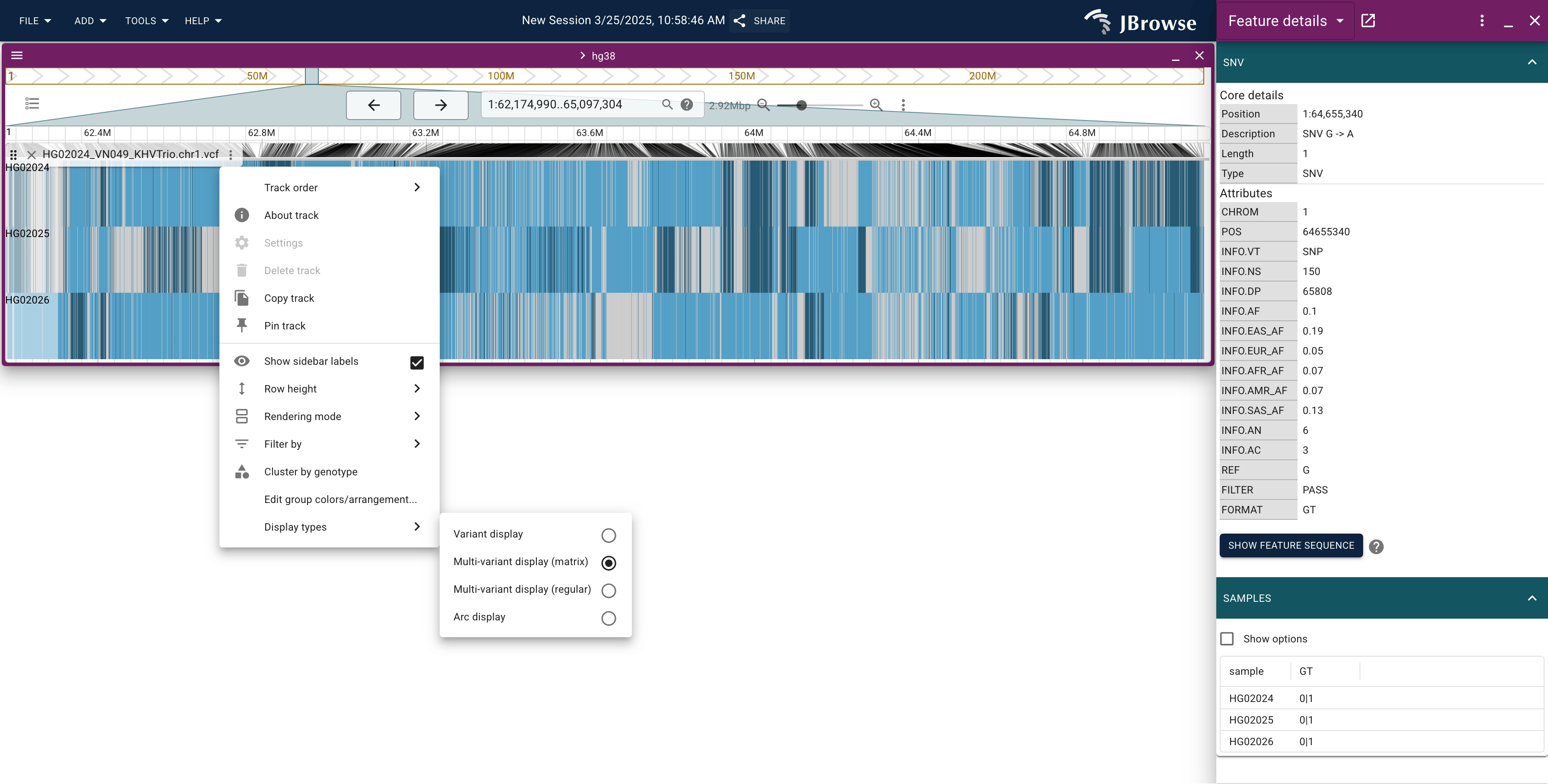
In the above figure, the default coloring for this matrix view is enabled.
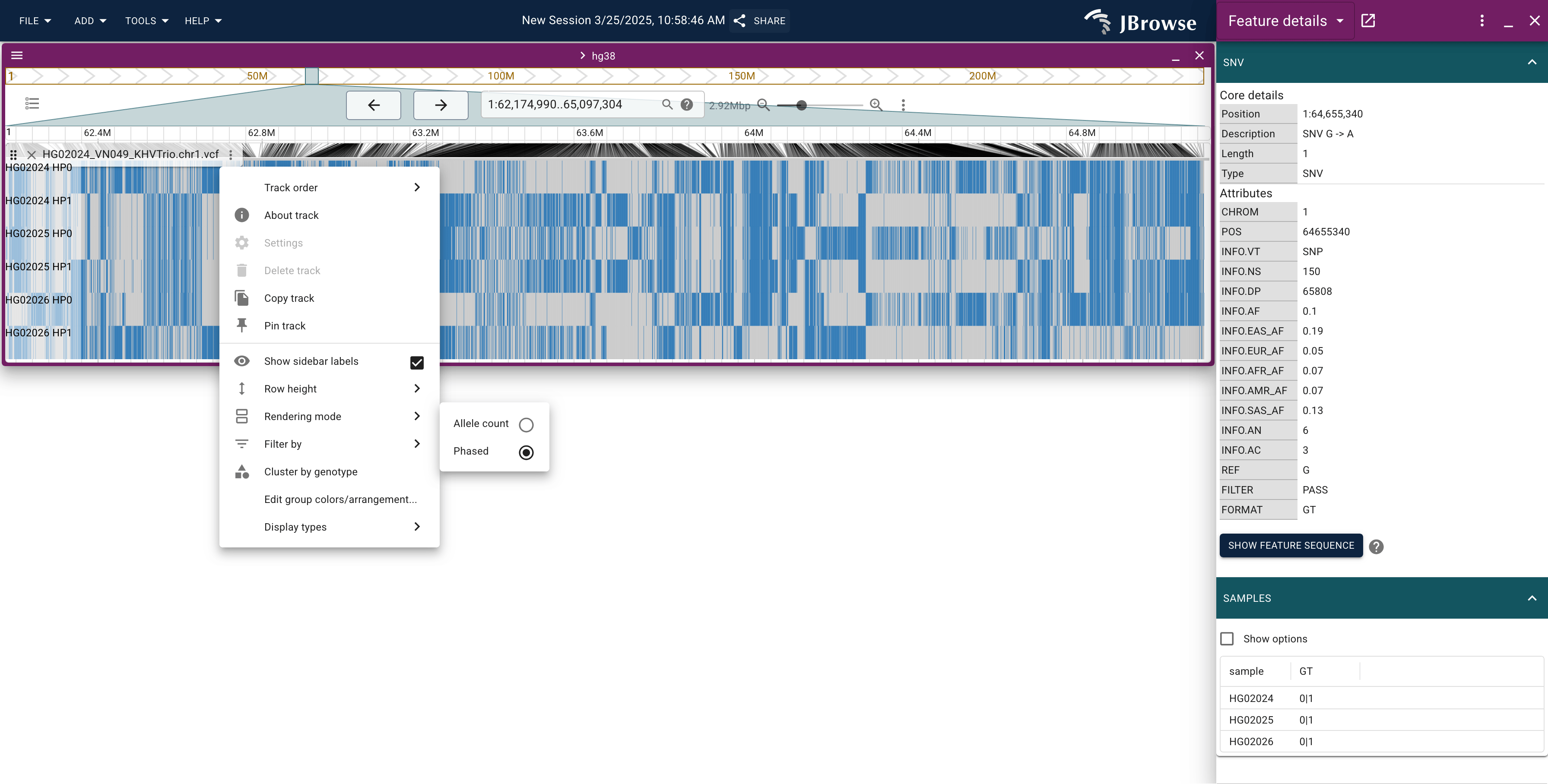
Enabling the "phased mode" of the matrix view
The matrix mode offers several options, one of which is the "phased" mode. If you have phased genotypes (e.g. you have genotypes with a vertical bar 0|1) for at least some of your variants, then this rendering mode will be available
The ideal is that your variants will be "completely phased". This sometimes requires specialized programs like SHAPEIT
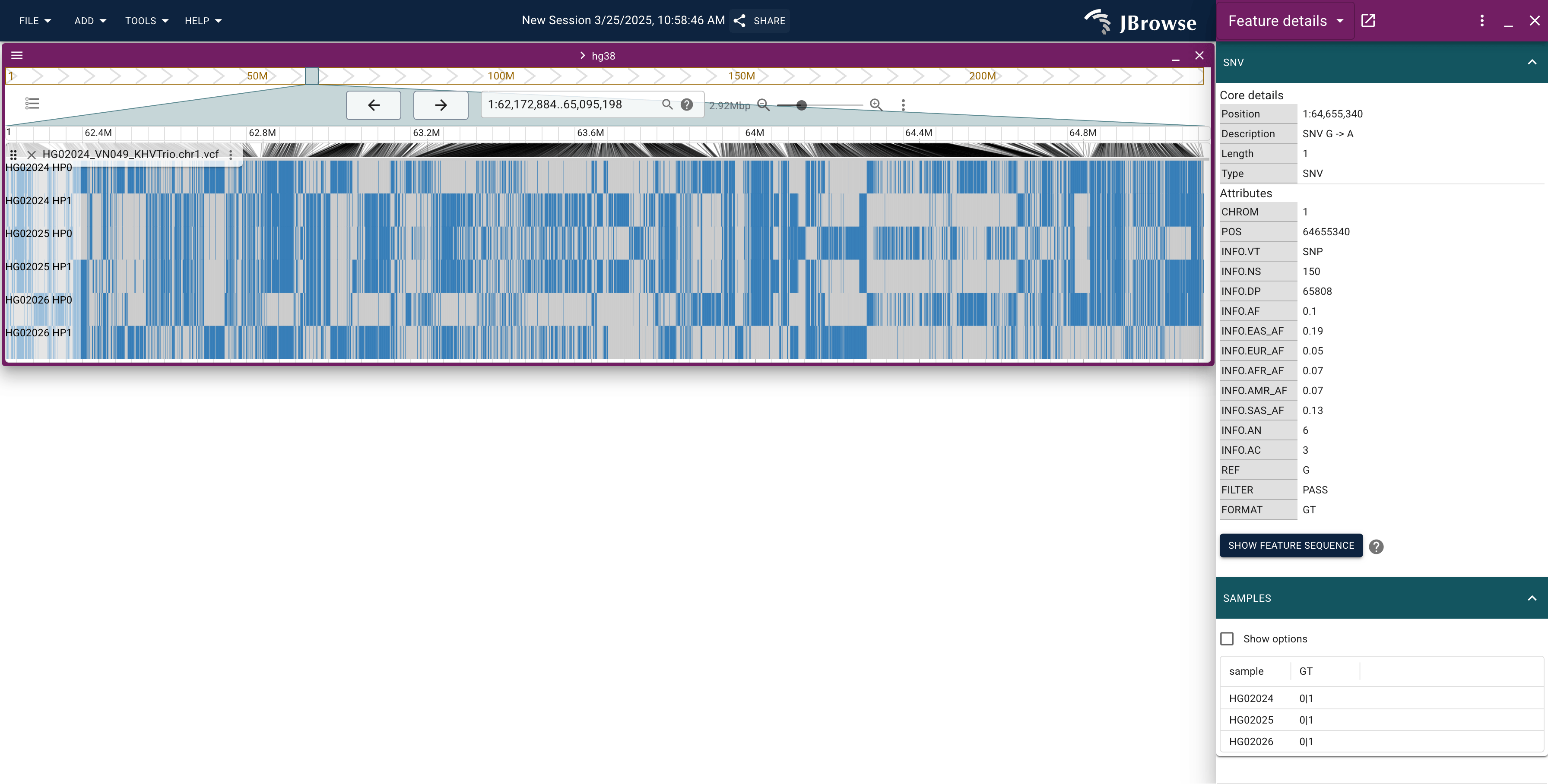
Finding matching haplotypes with "visual phasing"
The term "visual phasing" comes from the genetic genealogy subfield. I am borrowing it here, but the idea is simple: you can look at the genotype matrix here, and see areas where different rows are matching. You would expect that the child would match the mom in some places, and the dad in other places. And indeed, each row looks somewhat like a barcode, so you can find matching pieces like this
Using a program to help find phased blocks
In the above section, we could see matching blocks with our eyes, but can a program help?
There are indeed options for this. The program hap-ibd (https://github.com/browning-lab/hap-ibd) can find "identical by descent" blocks. It is actually capable of finding such blocks in massive population-scale datasets, but we are applying it here to a smaller trio VCF. The hap-ibd program takes as input:
- a phased VCF like the trio dataset (https://hgdownload.soe.ucsc.edu/gbdb/hg38/1000Genomes/trio/HG02024_VN049_KHV/HG02024_VN049_KHVTrio.chr1.vcf.gz)
- a genetic map in PLINK format (the README of hap-ibd provides these for GRCh38)
Converting hap-ibd data into a format for JBrowse
By loading the hap-ibd data in JBrowse, hap-ibd can tell us which blocks of the chromosome are matches
#!/bin/bash
# create a bed file from hap-ibd output with columns "chr, start, end, sample_name1 hap1 sample_name2 hap2"
# add this config to jbrowse get(feature,'sample1')+':HP'+get(feature,'hap1')+' '+get(feature,'sample2')+':HP'+get(feature,'hap2')
zcat result.ibd.gz | cut -f 5,6,7 >! out.bed
zcat result.ibd.gz | cut -f 1,2,3,4 >! samples.txt
echo "#chr start end sample1 hap1 sample2 hap2" > out.bed
paste out.bed samples.txt >> out.bed
After this conversion, we can load this simple BED file into JBrowse via the GUI or the CLI. It is probably small enough that it doesn't even need tabix conversion
Background: relationship between phased blocks, and the biology of recombination
Many people have heard of the term "recombination" or "crossing over" with regards to DNA, but what is it?
NHGRI "Crossing over, as related to genetics and genomics, refers to the exchange of DNA between paired homologous chromosomes (one from each parent) that occurs during the development of egg and sperm cells (meiosis)." (https://www.genome.gov/genetics-glossary/Crossing-Over)
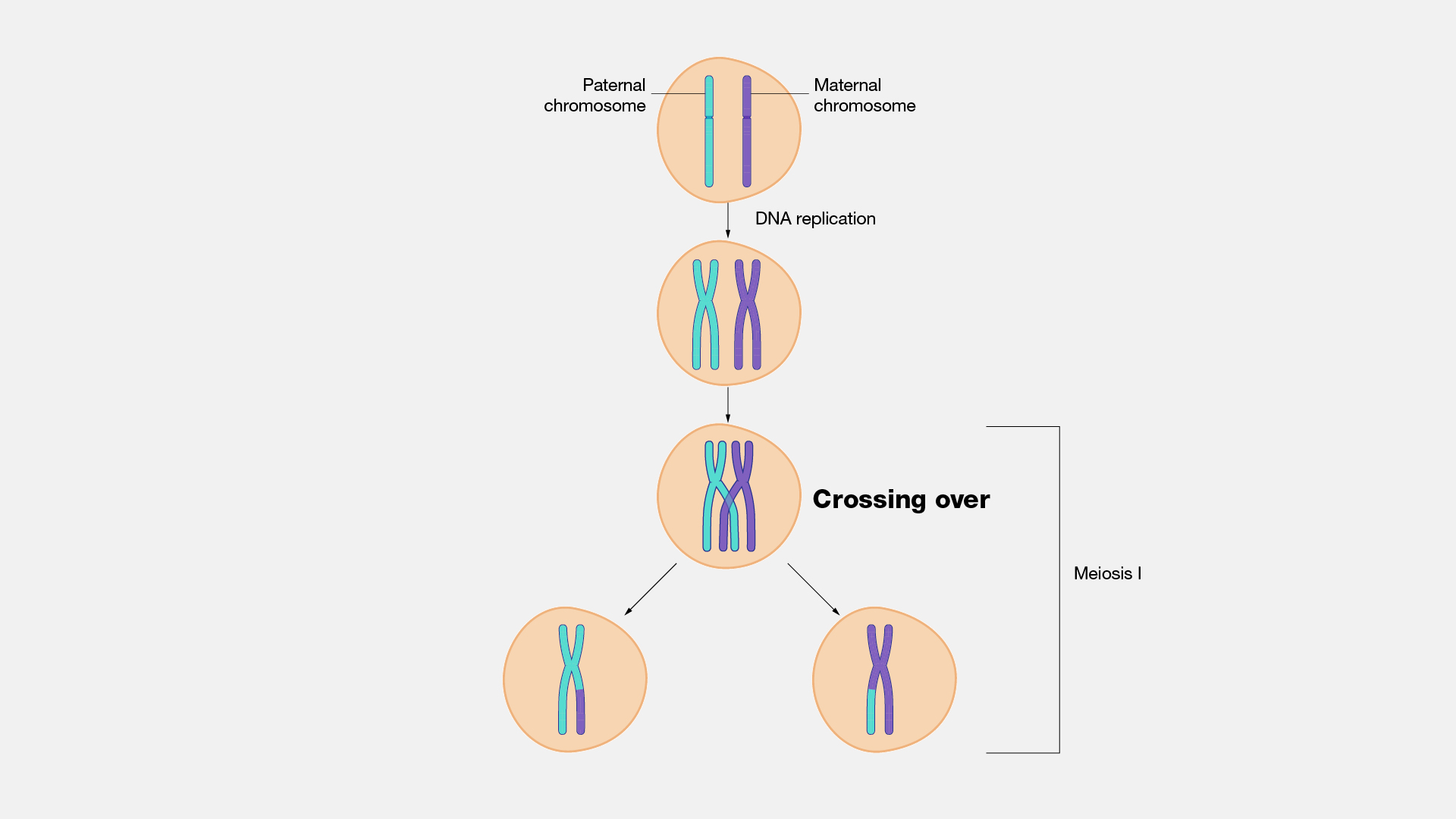 Figure from
https://www.genome.gov/genetics-glossary/Crossing-Over
Figure from
https://www.genome.gov/genetics-glossary/Crossing-Over
But there is a subtle but important point here: Your parents' genomes don't recombine during fertilization. Recombination happens during the production of their gametes. Therefore, your grandparents' genomes recombine during the production your parents sperm/eggs. We will see this visually below
Visualizing phased blocks and crossing over points in phased VCF files in JBrowse
After loading the hap-ibd track, we can see the blocks that hap-ibd calculated. We can additionally connect the lines onto the matrix view (which admittedly isn't straightforward, we have to follow the lines from the genomic position to the matrix position),
We can see this in the trio dataset where the child has a mixture of the dad's haplotypes and a mixture of the mom's haplotypes
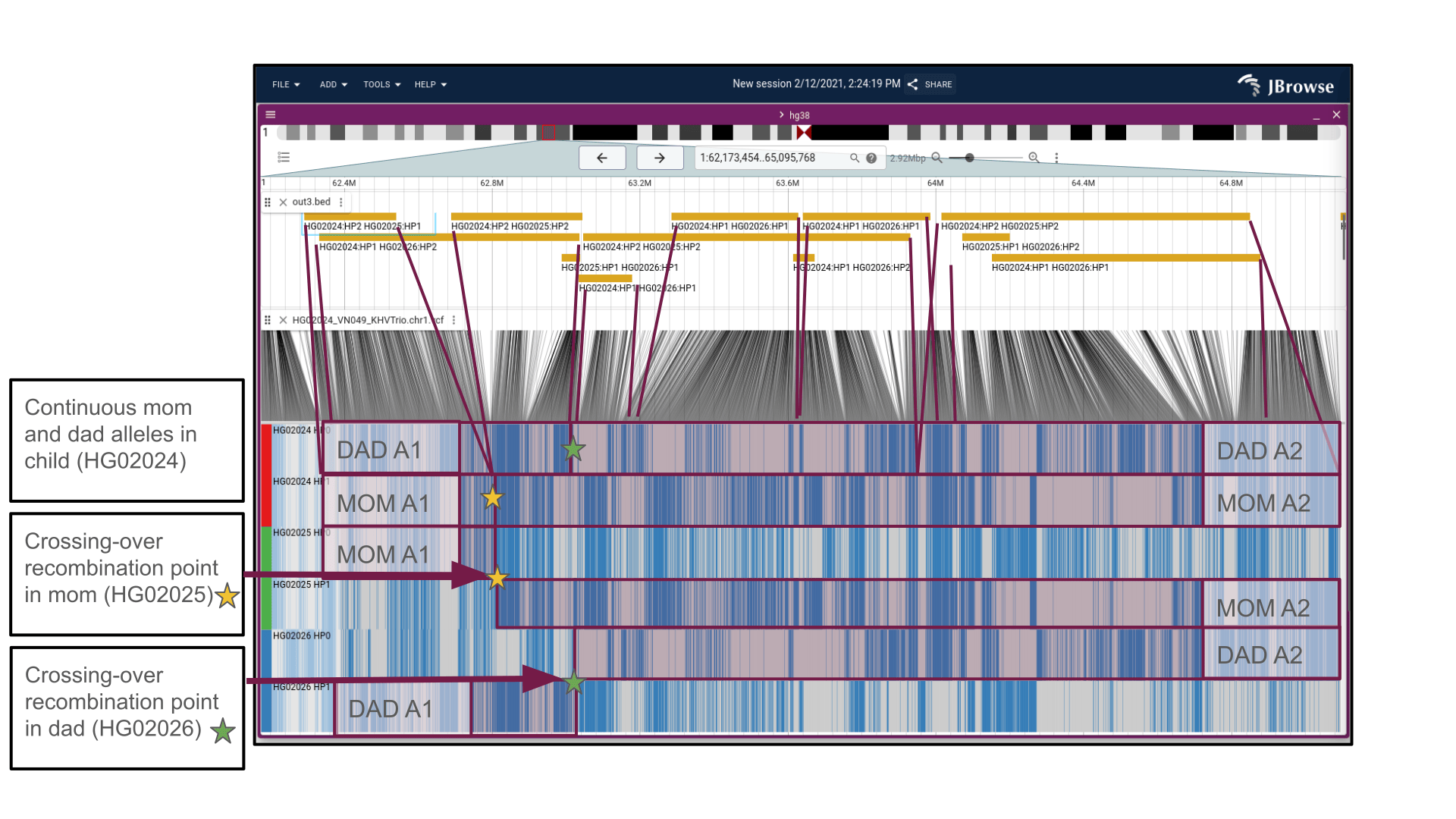
In the above screenshot, you can look at the 'barcode-like' patterns to see the matches between MOM A1 (allele 1) in mom and the child, MOM A2 (allele 2) in mom and child, DAD A1 (allele 1) in dad and child, DAD A2 (allele 2) in dad and child
You can see why we mentioned the grandparents above: for instance, the DAD A1 and DAD A2 alleles come from the crossing over of his two copies of his chromosomes that he got from his parents, which then recombine into a single chromosome in his child!
Footnote
The above procedure, particularly the final visualization with marked-up blocks, was done manually. It may be able to be improved in the future!
Author note: I found it quite wonderful to see this type of visualization! Feel free to let me know if I missed anything or if you have any other feedback