Visualizing DNA methylation
JBrowse 2 supports two complementary views of DNA methylation: per-read modification coloring directly on BAM/CRAM alignments, and aggregate methylation from bedMethyl files produced by tools like modkit. This tutorial demonstrates both using publicly available COLO829 melanoma ONT data and a chr20 nanopore methylation dataset.
Per-read methylation with BAM/CRAM
When a BAM or CRAM file carries base modification tags (MM/ML as specified in the SAM format specification), JBrowse 2 can color individual bases on each read by their modification probability. This works out of the box — no extra config is needed.
Coloring is enabled via Track menu → Pileup settings → Color by → Modifications or methylation. Each modification type gets a distinct color; for 5mC/5hmC data the typical default is red (5mC) and blue (5hmC). The color intensity reflects the modification probability (ML tag value).
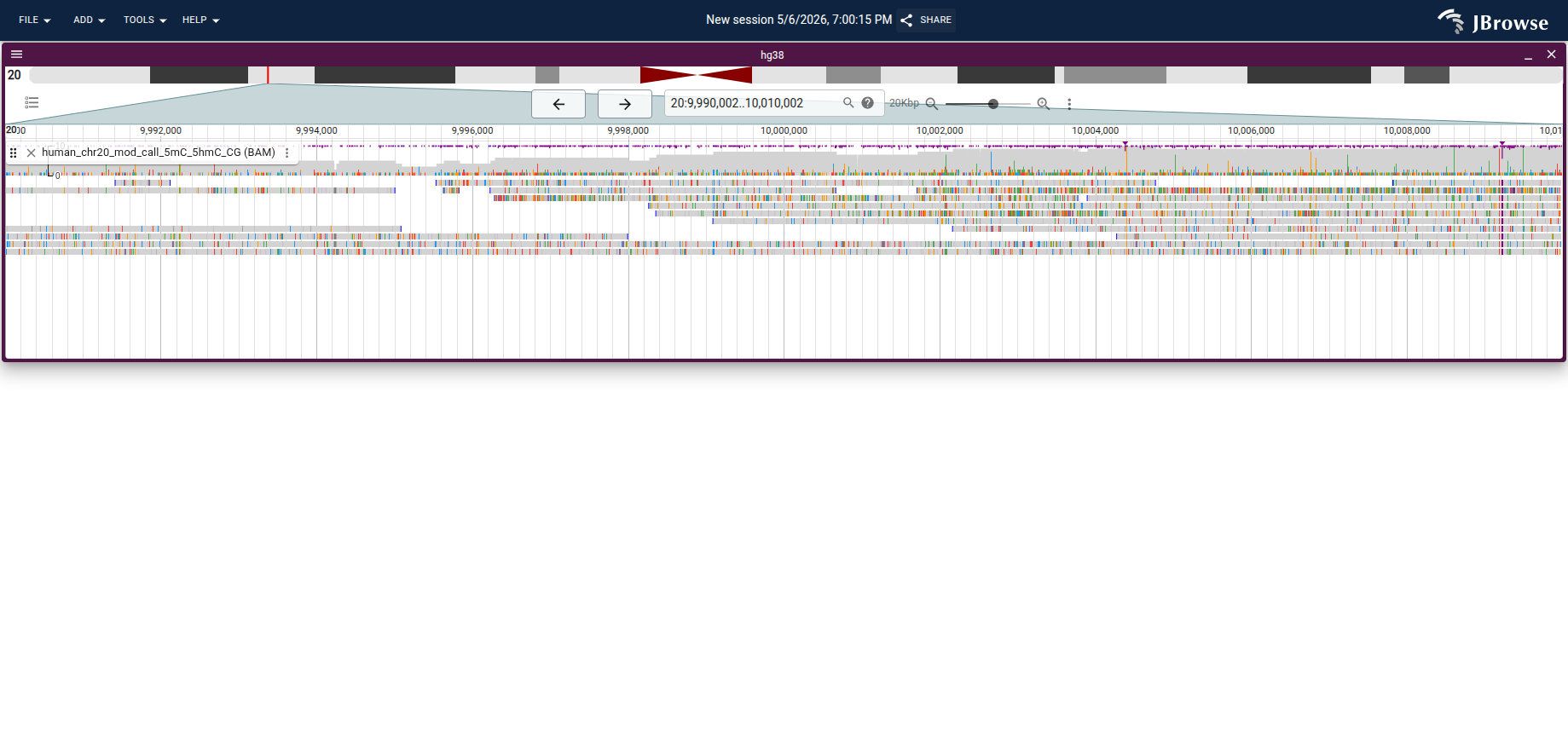
Live demo — chr20 per-read modification coloring
Aggregate methylation with modkit bedMethyl
modkit pileup aggregates per-read modification calls into a bedMethyl file — one row per CpG per modification type, with the fraction of reads carrying that modification. This is a compact format for storing population-level methylation across a whole genome.
Generating the file
# Standard (single modification fraction per CpG)
modkit pileup sample.bam output.bedmethyl --ref reference.fa --preset traditional
bgzip output.bedmethyl
tabix -p bed output.bedmethyl.gz
# Phased (produces hp1.bedmethyl, hp2.bedmethyl, combined.bedmethyl)
modkit pileup sample.bam output_dir/ --ref reference.fa --phased
bgzip output_dir/combined.bedmethyl
tabix -p bed output_dir/combined.bedmethyl.gz
--preset traditional collapses 5mC and 5hmC into a single 5mC fraction
(bisulfite-equivalent). Omit it to keep separate rows for each modification type
(m for 5mC, h for 5hmC).
Loading as a MultiQuantitativeTrack
Because bedMethyl is a BED-format file with a numeric score column, it can be
loaded using a BedTabixAdapter inside a MultiQuantitativeTrack. JBrowse
reads the modification type from the name column (e.g. m for 5mC, h for
5hmC) and creates one subtrack per type.
{
"type": "MultiQuantitativeTrack",
"trackId": "sample_modkit",
"name": "CpG methylation (modkit)",
"assemblyNames": ["hg38"],
"adapter": {
"type": "BedTabixAdapter",
"bedGzLocation": {
"uri": "https://yourhost/sample_modkit.bed.gz"
},
"index": {
"location": {
"uri": "https://yourhost/sample_modkit.bed.gz.tbi"
}
}
}
}
The Y-axis shows the percent methylation (0–100). Each CpG position appears as a
vertical bar; the two subtracks (h for 5hmC and m for 5mC) are stacked in
multirow mode by default so their scales are independent.
Example: COLO829 tumor with CRAM and bedMethyl
The screenshot below shows the COLO829 melanoma tumor ONT dataset at
chr20:10,000,000–10,003,000. The upper track is the CRAM alignment with reads
colored by modification; the lower track is the modkit bedMethyl file loaded as
a MultiQuantitativeTrack with h (5hmC) and m (5mC) subtracks.
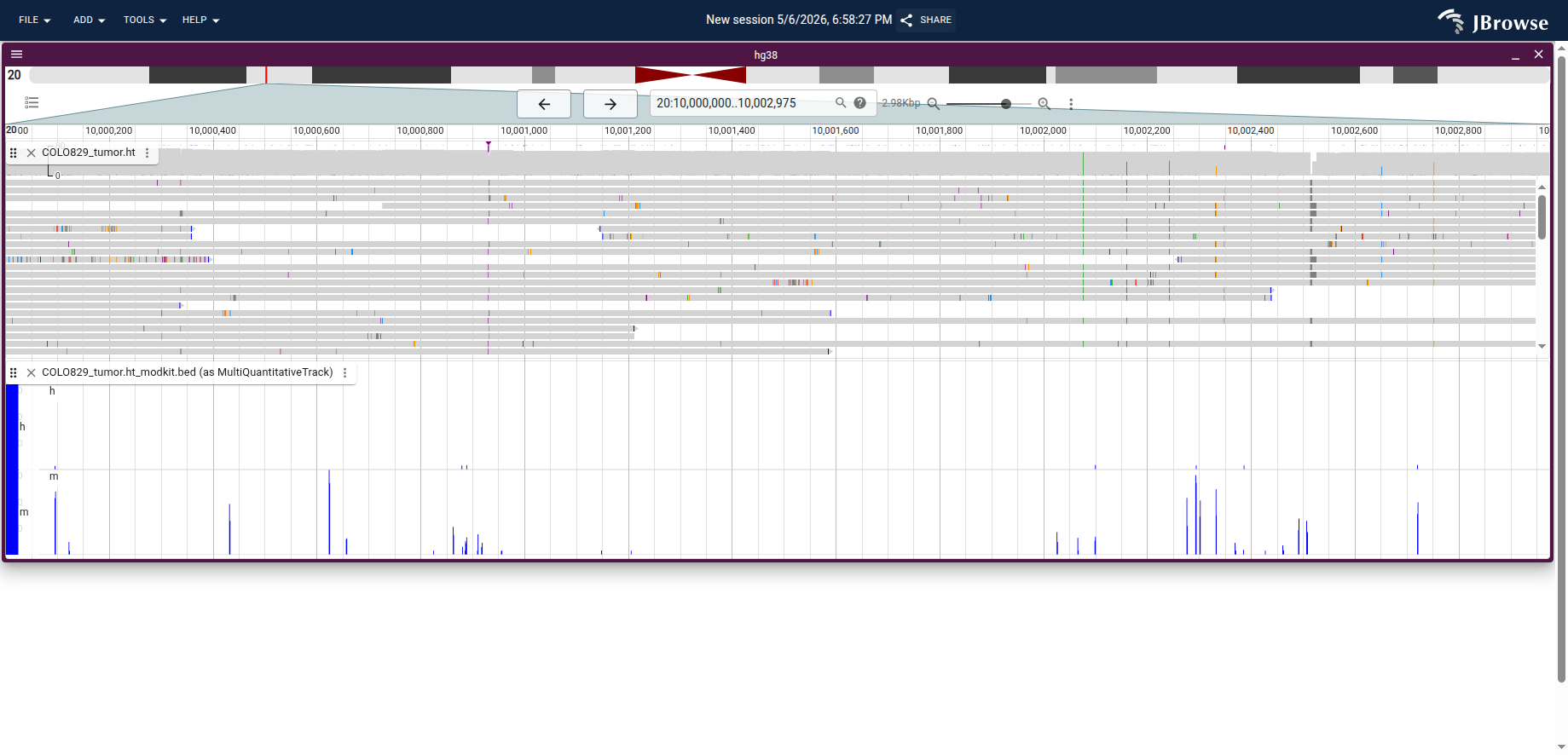
Live demo — COLO829 CRAM + bedMethyl
Choosing between the two approaches
| Approach | Best for | | -------------------------------- | -------------------------------------------------------------------------------------------- | | Per-read BAM/CRAM coloring | Haplotype-aware methylation, allele-specific methylation, individual read inspection | | bedMethyl MultiQuantitativeTrack | Whole-genome methylation overview, comparing tumor vs normal, fast loading at any zoom level |
For haplotype-aware analysis, combine both: load a haplotagged BAM (with HP tags from WhatsHap or HiPhase), color by modification, and sort by HP tag to see per-haplotype methylation patterns. The bedMethyl track provides the aggregate signal for quick navigation to regions of interest.